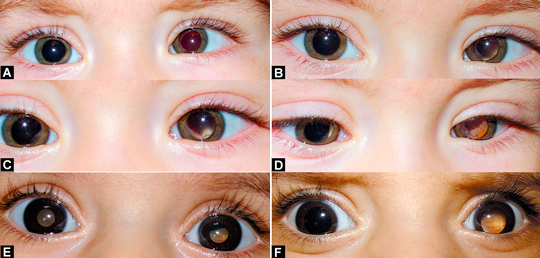
4-30 A. Bilateral retinoblastom hastasında sol gözde içe şaşılık ve homojen kırmızı yansıma izleniyor (sol gözde çok geniş, sağda ise sadece nazal bölgede sınırlı lezyon mevcut). B. Aynı hasta biraz farklı bir yöne bakarken sol gözdeki kırmızı yansımada bir problem olduğu dikkati çekiyor. C. Hasta kitlenin bulunduğu bölgeye doğru bakarken solda lökokori belirginleşiyor. D. Sağ göz nazalinde de lökokorinin olduğu izleniyor. E-F. Başka bir hastada solda daha büyük lezyonun olduğu bilateral
retinoblastoma bağlı lökokori.
Retinoblastom olgularının büyük bir kısmı indirek oftalmoskopi yardımı ile oftalmolog tarafından tanınabilir. Kalsifikasyonların belirlenmesinde BT ve ultrason, MRG ise ek olarak tümör boyutlarının ve yayılımının belirlenmesinde faydalı görüntüleme yöntemleridir. Küçük bir grup hastada ise tüm bu tetkik ve muayenelere rağmen lökokori yapan diğer nedenlerden ayırt edilmesi oldukça zorluk taşıyabilir.
Uluslararası intraoküler retinoblastom sınıflaması (2005);
• Grup A: çok düşük risk (kritik göz yapılarından uzakta küçük ayrık tümörler)
• Grup B: düşük risk (vitreusa veya retina altına tomurcuklanma göstermiyor)
• Grup C: orta risk (herhangi boyutta fokal vitreus veya retina altına tomurcuklanma)
• Grup D: yüksek risk (yaygın tomurcuklanma, retinanın 1/4’ünde dekolman)
• Grup E: enükleasyon (lense yaslanan tümör, neovasküler glokom, ön segmentte yaygın infiltrasyon, büyük intraoküler hemoraji, fitizis ve aseptik orbita selüliti)
Evrelendirme (Chantada 2006);
• Evre 0: Grup A, B, C, D
• Evre 1: Grup D, E (enükleasyon yapılmış olan gözde tümör histolojik olarak tamamen çıkarılmış)
• Evre 2: Grup E (enükleasyon yapılmış olan gözde histolojik olarak koroid veya retrolaminar bölgede yoğun yayılım var)
• Evre 3: Orbita yayılımı, preaurikular veya servikal lenfadenopati
• Evre 4: Hematojenik (kemik iliği) ve santral sinir sistemine metastaz
Tedavi
Retinoblastom tedavisinin belirlenmesinde tümörün büyüklüğü, yerleşim yeri, unilateral veya bilateral olması, orbita invazyonu ve metastaz varlığı önemli faktörler olarak öne çıkmaktadır. Tedavi planı belirlenmeden önce çocuğun genel anestezi altında detaylı fundus muayenesinin yapılıp, lezyonların çizimler ve fotoğraflar vasıtasıyla dökümantasyonunun yapılması gerekir. Tedavi ve takip süreci oftalmolog ve pediatrik onkologdan oluşmuş bir ekip çalışması gerektirir.